


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
 system
system黒崎 譲; 高柳 敏幸*
Chemical Physics Letters, 406(1-3), p.121 - 125, 2005/04
BrH系の非経験的に計算された基底状態のグローバルなポテンシャルエネルギー面(PES)についての新しい解析的関数を構築した。これは、以前われわれが発表した1 A' PES[Y. Kurosaki, T. Takayanagi, J. Chem. Phys. 119 (2003) 7383]の修正版である。反応H+HBr
A' PES[Y. Kurosaki, T. Takayanagi, J. Chem. Phys. 119 (2003) 7383]の修正版である。反応H+HBr H+Brとその同位体置換した反応の速度定数を、新しい1A' PESを用いて計算したところ実測値をよく再現した。これはフィットした関数の反応障壁の値1.53kcal mol
H+Brとその同位体置換した反応の速度定数を、新しい1A' PESを用いて計算したところ実測値をよく再現した。これはフィットした関数の反応障壁の値1.53kcal mol が真の値に非常に近いことを強く示唆している。
が真の値に非常に近いことを強く示唆している。
 potential energy surfaces for the lowest three doublet states (1A', 2A', and 1A") of the BrH system
potential energy surfaces for the lowest three doublet states (1A', 2A', and 1A") of the BrH system黒崎 譲; 高柳 敏幸
Journal of Chemical Physics, 119(15), p.7838 - 7856, 2003/10
被引用回数:26 パーセンタイル:63.68(Chemistry, Physical)BrH系の三つの最低二重項状態(1A', 2A', 1A") についての断熱ポテンシャルエネルギー面を、Breit-Pauliハミルトニアンに基づくスピン-軌道相互作用による補正を加えてMRCI/aug-cc-pVTZ法により計算し、得られた断熱エネルギーを解析的な多体関数にフィットした。基底状態のポテンシャル面上での水素引抜き及び水素交換の反応障壁は、MRCI+Q/aug-cc-pVTZレベルでそれぞれ1.28 and 11.71 kcal molと計算された。ポテンシャル面のフィットの精度は0.1 kcal mol以内であった。フィットした基底状態のポテンシャル面を用いて、水素引抜き及び水素交換並びに同位体置換した反応の反応速度定数を、変分的遷移状態理論に基づくICVT/LAG法により計算した。その結果、水素引抜きについては実験との一致はおおむね良好であったが、水素交換については計算値は実験値を大幅に下回った。この不一致は、実験データの不足が主な原因と考えられる。
大和田 謙; 鈴木 和弥
Spectrochimica Acta, Part A, 50(6), p.1057 - 1063, 1994/00
赤外・ラマンスペクトルデータの基準振動解析から得られる二次のポテンシャル定数(力の定数)を応用して、多原子分子形成時の電子化学ポテンシャルを求める方法を検討した。本報では密度汎関数理論を採用して、これに単純結合一電荷モデル(Simple Bond-Charge Model)を試験的に組み込んだ近似法を提案した。この方法を用いて、種々の異核二原子分子及び多原子分子の電子化学ポテンシャルを計算し、実験値及びab-initio SCF計算から得られる値と比較した結果、本法の有用性を確かめることができた。
大和田 謙
Spectrochimica Acta, Part A, 49(1), p.81 - 94, 1993/00
異核=原子分子の基準振動解析から得られる調和および非調和ポテンシャル定数を応用して、分子の化学ポテンシャルを求める方法を検討した。本法は主として密度汎関数理論に基礎を置き、分子中の各原子のエネルギーが電子数と核ポテンシャルに関して展開できることを利用する。これらの展開は、N個の電子数と与えられた核ポテンシャルv(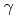 )をもつ系の基底状態の全エネルギーがN,v()の汎関数として表されるので可能となる。本法を用いて、多数のアルカリハライド分子、その他の異核=原子分子の化学ポテンシャルを計算し、Sandersonの原理及びab-initio SCF計算から得られる値と比較した結果、本法の有用性を確かめることができた。また見掛けの化学ハードニス及び福井関数を含む積分項の値を評価した。分子形成時の化学ポテンシャル変化についても簡単に議論した。
)をもつ系の基底状態の全エネルギーがN,v()の汎関数として表されるので可能となる。本法を用いて、多数のアルカリハライド分子、その他の異核=原子分子の化学ポテンシャルを計算し、Sandersonの原理及びab-initio SCF計算から得られる値と比較した結果、本法の有用性を確かめることができた。また見掛けの化学ハードニス及び福井関数を含む積分項の値を評価した。分子形成時の化学ポテンシャル変化についても簡単に議論した。
大和田 謙
Journal of Physical Chemistry, 96(14), p.5825 - 5829, 1992/00
被引用回数:2 パーセンタイル:11.73(Chemistry, Physical)異核二原子分子の基準振動解析から得られる調和および非調和ポテンシャル定数を応用して、分子形成時の各原子の化学ポテンシャル変化と分子の化学ポテンシャルを求める方法を検討した。本法は主として密度汎関数理論に基礎を置き、分子中の各原子のエネルギーが電子数と核ポテンシャルに関して展開できることを利用する。これらの展開は、N個の電子数と与えられた核ポテンシャルv(F)をもつ系の基底状態の全エネルギーがN,v(F)の汎関数として表されるので可能となる。本法を用いて、多数のアルカリハライド分子、その他の異核二原子分子の原子および分子の化学ポテンシャルを計算し、Sandersonの原理から得られる値と比較した結果、本法の有用性を確かめることができた。
大和田 謙
Spectrochimica Acta, Part A, 47(12), p.1751 - 1765, 1991/00
累核二原子分子の基準振動解析から得られる調和および非調和ポテンシャル定数を応用して、分子形成時の移動電子数(電荷移動)と電気双極子モーメント変化(関数)を求める方法を検討した。本法は主としてスレーターの軌道展開法に基礎を置き、電子スピンを無視する近似において、分子中の各原子エネルギーが各原子軌道を占有する電子数で展開できるということを利用する。本法の精度と信頼性を確かめるために、多数のアルカリハライド分子、その他の累核二原子分子を例にとり、それらの移動電子数と電気双極子モーメントを計算し、実測値と比較した。特に現在最も信頼しうるHF、OH分子の実測電気双極モーメント関数と詳細な比較を行った結果、本法の有用性を確かめることができた。
大和田 謙
Spectrochimica Acta, Part A, 46(10), p.1461 - 1473, 1990/00
2原子分子の基準振動の解析から得られる調和及び非調和ポテンシャル定数(カの定数)を応用して、分子のエネルギー成分(電子運動エネルギー、全静電ポテンシャルエネルギー、電子、核引力エネルギー、電子間反発エネルギー、核間反発エネルギー、Hartree-Fock固有値)を経験的に求める方法を検討した。本研究で開発した方法は、量子力学的ヴィリアル定理から導びかれる非同次線形3階微分方程式及び原子番号に関する分子エネルギーの同次性の仮説に基礎をおいている。本法の精度は、精密計算によって得られているHartree-Fockデータとの比較によって確かめられ、これによって、本法は上記の原子間距離依存分子エネルギー成分を求めるのに適していることがわかった。
大和田 謙
Spectrochimica Acta, Part A, 45(4), p.487 - 490, 1989/00
2原子分子のポテンシャル定数を用いて、分子のエネルギー成分(電子運動エネルギー、静電ポテンシャルエネルギー等)を経験的に求める方法を検討した。量子力学的ウィリアル定理から運動エネルギー表現および静電ポテンシャルエネルギー表現の非同次線型1階微分方程式を導き、これらの解から分子エネルギー成分を経験的に算出する方法を見出した。本法の精度は、精密計算によって得られているHartree-Fockデータとの比較によって確かめられ、これによって、本法は原子間距離依存の分子エネルギー成分を求めるのに適していることがわかった。
大和田 謙
Journal of Chemical Physics, 85(10), p.5882 - 5889, 1986/10
被引用回数:3 パーセンタイル:18.02(Chemistry, Physical)ParrおよびGadreによって報告された全分子エネルギーと電子エネルギーの核電荷に関する2つのエネルギー同次性の仮定を考察して、さらに一歩進んだ(両仮定の欠点を取除いた)全分子エネルギーに関するエネルギー同次性の仮定を提案した。この仮定は全分子エネルギーを局在電子エネルギーと原子間距離に依存する非局在エネルギーとに分離し、その各々についてエネルギー同次性を考慮したのもである。この仮定の妥当性を検討した結果、分子の種々の性質(分子エネルギー成分、分子内ポテンシャル系、高次のポテンシャル定数など)を記述するのに適したものであることがわかった。また、この仮定をHartree-Fock法に組み入れた場合の効果についても議論した。
大和田 謙
Journal of Chemical Physics, 72(1), p.1 - 6, 1980/00
被引用回数:41 パーセンタイル:79.96(Chemistry, Physical)等核二原子分子に摂動論を応用して得られる力の定数を用いて、種々の原子の有効核電荷が定義された。これらの有効核電荷は、直接、異核二原子分子の力の定数の計算に転用できることが理論的に証明された。上で定義された有効核電荷を多原子分子へ応用するため、静電理論にもとづく有効分子内ポテンシャル関数が提案され、これによって、まず、三原子分子の力の定数が計算された。これらは実測振動数から得られた値と比較的良く一致することが分った。したがって、有効核電荷を用いる本法は多原子分子の力の定数の推定に極めて有用であると思われる。
大和田 謙
Chemical Physics Letters, 66(1), p.149 - 153, 1979/00
被引用回数:1有効核電荷を含む静電ポテンシャル関数にもとづいて多原子分子の変角の力の定数、Hjk、を次式のように導いた。 Hjk=Zj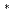 Zk/qjk
Zk/qjk (SjikSkij+2TjikTkij) ここにqは平衡位置における非結合原子間距離、Zは有効核電荷、SおよびTは分子の幾何学的パラメータである。上式を多くの三原子分子を用いて評価した結果、変角の力の定数の推定に有用であることがわかった。
(SjikSkij+2TjikTkij) ここにqは平衡位置における非結合原子間距離、Zは有効核電荷、SおよびTは分子の幾何学的パラメータである。上式を多くの三原子分子を用いて評価した結果、変角の力の定数の推定に有用であることがわかった。
